Developmental Biology: The Blueprint of Human Identity
- Introduction and Historical Context
- Embryological Origin and Early Development
- Sexual Differentiation: The Role of AMH
- Fate in the Female: Differentiation and Derivatives
- Fate in the Male: Regression and Vestigial Remnants
- Clinical Significance: Müllerian Duct Anomalies (MDA)
- Specific Müllerian Duct Anomalies
- Diagnostic Techniques and Management
- Genetic and Hormonal Regulation
Introduction and Historical Context
The Müllerian ducts, also known scientifically as the paramesonephric ducts, represent crucial paired embryological structures that are fundamentally present in all developing human and mammalian embryos regardless of genetic sex in the initial stages of gestation. These structures were first meticulously described and characterized by the renowned German anatomist and physiologist, Johannes Müller (1801–1858). Müller’s seminal work explained how these ducts serve as the biological blueprint for the eventual development of the internal female reproductive system, marking a critical step in understanding sexual differentiation. His contributions established the foundational knowledge necessary to distinguish between the male and female developmental pathways during the embryonic period, setting the stage for future research into reproductive anatomy and endocrinology.
The enduring significance of Müller’s findings lies in the recognition that the female reproductive system represents the default developmental pathway in the absence of specific inhibitory hormonal signals. The presence of the Müllerian ducts dictates the potential for forming the fallopian tubes, the uterus, the cervix, and the upper segment of the vagina. Conversely, in the male embryo, these ducts must actively regress and disappear, a process facilitated by potent hormones synthesized by the developing testes. This duality highlights a complex regulatory mechanism where the persistence or regression of the ducts is the primary determinant of internal sexual anatomy, demonstrating the critical transitional phase that occurs between the indifferent state and definitive sexual differentiation.
Historically, the study of the paramesonephric system provided the first clear insight into the concept of bipotentiality in the early embryo, where two parallel duct systems—the Wolffian (mesonephric) ducts and the Müllerian ducts—coexist temporarily. It is the subsequent hormonal environment that determines which system persists and differentiates, and which system disintegrates. The formal tone adopted in the anatomical literature reflects the precision required when discussing these sensitive developmental processes, which are prone to complex malformations if the delicate hormonal signaling cascade is disrupted.
Embryological Origin and Early Development
The genesis of the Müllerian ducts begins early in the human embryo, typically around the sixth week of gestation. They arise from an invagination of the coelomic epithelium, specifically adjacent to the cranial end of the mesonephric (Wolffian) duct system within the intermediate mesoderm. This proximity is critical, as the development and eventual fate of the Müllerian ducts are intrinsically linked to the presence and regression of the Wolffian ducts, which serve as the foundation for the male reproductive tract. Initially, the paramesonephric ducts appear as paired funnels opening into the coelomic cavity near the developing kidneys, extending caudally and medially towards the midline of the body.
As development progresses, the caudal tips of the two ducts migrate toward the midline. They eventually cross the Wolffian ducts ventrally and fuse together, forming a single, median structure known as the uterovaginal canal. This fusion event is pivotal for the formation of the uterus and the upper vagina in female embryos. The unfused cranial portions of the ducts remain distinct, forming the paired fallopian tubes. This period, characterized by the simultaneous presence of both the Müllerian and Wolffian systems, is referred to as the indifferent stage, lasting until the influence of genetically determined sex hormones begins to exert its differential effect on the duct systems.
The proper formation and alignment of the Müllerian ducts are dependent on complex cellular signaling pathways and precise migratory patterns. Any perturbation in the development of the intermediate mesoderm or the coelomic epithelium during this early phase can lead to significant congenital anomalies later in life. The precise timing of the canalization and fusion processes is regulated by various transcription factors and growth factors, ensuring that the structures are appropriately positioned relative to the developing urinary tract and the adjacent pelvic organs, necessitating a high level of coordinated growth within the embryonic environment.
Sexual Differentiation: The Role of AMH
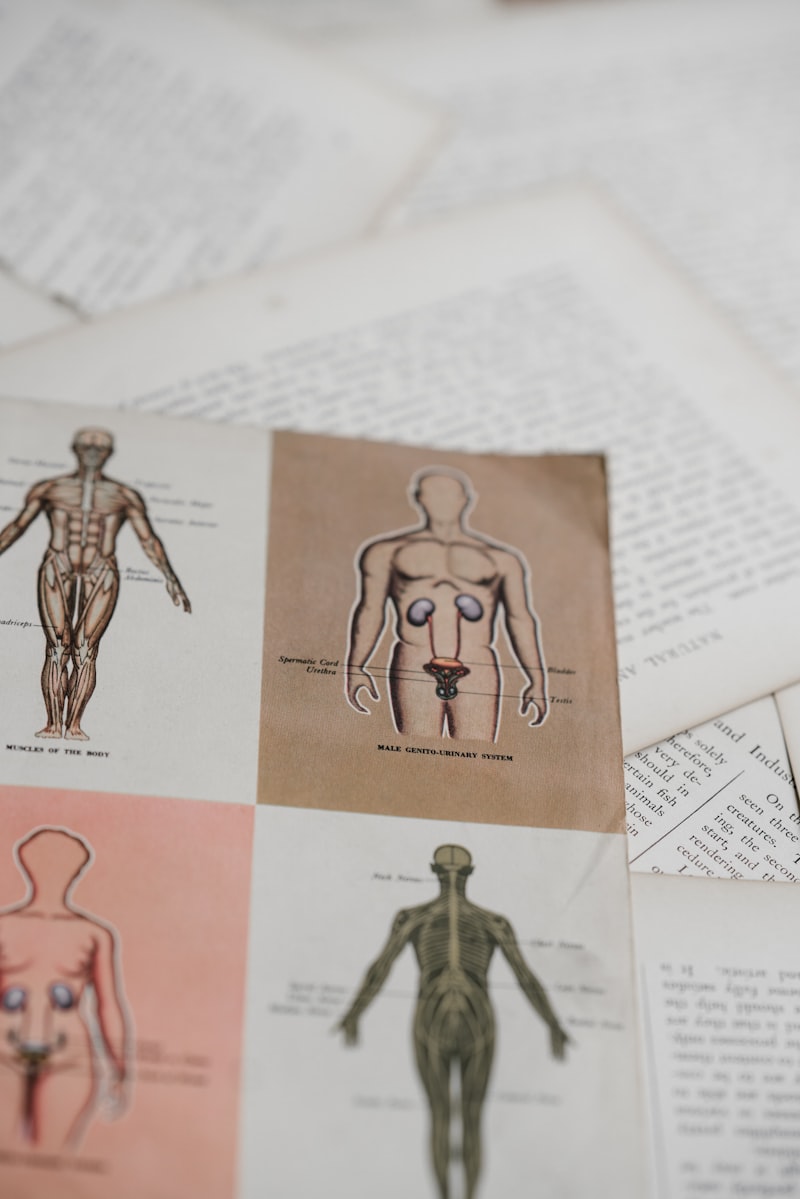
The critical transition from the bipotential, indifferent state to definitive sexual anatomy hinges upon the presence or absence of a specific hormonal signal: Anti-Müllerian Hormone (AMH), also known as Müllerian-Inhibiting Substance (MIS). This hormone is the primary factor responsible for determining the fate of the Müllerian ducts. In male embryos (XY genotype), the developing testes contain Sertoli cells, which begin producing large quantities of AMH starting around the seventh to eighth week of gestation. This powerful glycoprotein acts locally and systemically, inducing the rapid regression and subsequent disappearance of the Müllerian ducts.
The mechanism of AMH action involves binding to specific receptors (AMH Type II receptor) expressed on the mesenchymal cells surrounding the Müllerian duct epithelium. This binding initiates a cascade of intracellular events, primarily involving apoptosis (programmed cell death) and the degradation of the ductal tissue. The efficiency of this regression is paramount; incomplete regression in a male embryo can result in the persistence of vestigial remnants, such as the appendix testis or, in rare cases, a uterus-like structure, a condition often associated with Persistent Müllerian Duct Syndrome (PMDS).
Conversely, in female embryos (XX genotype), the ovaries do not produce significant levels of AMH during this critical developmental window. The absence of this inhibitory signal allows the Müllerian ducts to persist, proliferate, and differentiate into the female reproductive organs. Therefore, the female pathway is considered the default developmental trajectory, requiring only the lack of AMH for the Müllerian structures to flourish. This fundamental difference—active destruction in males versus passive persistence and differentiation in females—underscores the complexity of hormonal regulation in human embryogenesis.
Fate in the Female: Differentiation and Derivatives
When the Müllerian ducts are permitted to persist due to the lack of AMH signaling, they undergo extensive differentiation to form the entirety of the internal female reproductive tract, excluding the ovaries themselves. This process involves elongation, differentiation of the epithelial lining, and the crucial fusion of the lower segments. The derivatives of the Müllerian ducts are systematically categorized based on their original position within the developing structure.
The key derivatives resulting from the successful development and transformation of the Müllerian ducts include:
- The Fallopian Tubes (Oviducts): These structures develop from the superior, unfused cranial portions of the ducts. They remain separate and open into the peritoneal cavity, providing the pathway for ova transport.
- The Uterus and Cervix: These centralized organs are formed from the fusion of the caudal portions of the two ducts in the midline. The muscular wall (myometrium) and the inner glandular lining (endometrium) differentiate from the surrounding mesenchyme.
- The Upper Third of the Vagina: The superior segment of the vagina is also derived from the uterovaginal canal, which forms after the fusion of the Müllerian ducts and their subsequent interaction with the urogenital sinus.
The differentiation process must be tightly regulated to ensure patency, proper muscularization, and functional integrity. The epithelium lining these structures will eventually differentiate into the specific cell types required for reproductive function, such as ciliated cells in the fallopian tubes necessary for ovum transport and secretory cells in the endometrium crucial for implantation. Failures in any stage of this complex differentiation, especially the fusion process, lead directly to congenital uterine anomalies that significantly impact fertility and obstetric outcomes.
Fate in the Male: Regression and Vestigial Remnants
In the male embryo, the fate of the Müllerian ducts is one of necessary destruction, driven entirely by the production of AMH from the Sertoli cells of the fetal testes. This regression must be complete and timely to allow the mesonephric (Wolffian) ducts to develop unobstructed into the male internal accessory structures, including the epididymis, vas deferens, and seminal vesicles. If regression is incomplete, the presence of Müllerian remnants can interfere with the descent of the testes or cause obstruction.
Despite the comprehensive regression induced by AMH, small, non-functional vestigial remnants of the Müllerian ducts often persist in the adult male anatomy. These remnants are typically clinically insignificant but serve as anatomical markers of the duct’s developmental history. The key vestigial structures include:
- The Appendix Testis (Hydatid of Morgagni): A small, stalked appendage located on the superior pole of the testis or the head of the epididymis. It represents the cranial, unfused tip of the Müllerian duct.
- The Prostatic Utricle (Vaginal Utricle): A small blind pouch located within the prostatic urethra. This structure represents the fused caudal portion of the Müllerian ducts.
The clinical significance of these remnants is generally low, although the appendix testis can occasionally undergo torsion, presenting as acute scrotal pain (mimicking testicular torsion). However, the failure of complete regression, as seen in PMDS, often involves a defect in AMH production or receptor function, leading to the presence of a rudimentary uterus and fallopian tubes in a genetically male individual. This condition confirms the potent inhibitory role of AMH and illustrates the consequences of its deficiency in the XY environment.
Clinical Significance: Müllerian Duct Anomalies (MDA)
Müllerian Duct Anomalies (MDA), or congenital uterine anomalies (CUAs), represent a spectrum of reproductive tract abnormalities that arise from errors during the development, fusion, or resorption phases of the paramesonephric ducts. These anomalies can range from asymptomatic minor uterine variations to severe malformations, often leading to significant reproductive morbidity, including recurrent pregnancy loss, infertility, preterm labor, and obstructed menstruation (hematocolpos or hematometra).
The classification of these anomalies is essential for accurate diagnosis and management, with the American Society for Reproductive Medicine (ASRM) system being the most widely accepted standard, categorizing defects into classes I through VII. These classifications are critical because they dictate the appropriate surgical intervention, such as hysteroscopic metroplasty for a septate uterus, or reconstructive surgery for a bicornuate uterus. Understanding the specific embryological defect—whether a failure of formation, fusion, or septal resorption—is paramount for predicting potential functional outcomes.
One of the most profound anomalies is Mayer-Rokitansky-Küster-Hauser (MRKH) Syndrome (ASRM Class I), characterized by the congenital absence or severe hypoplasia of the uterus and upper vagina, resulting from a failure of the Müllerian ducts to develop fully. Although individuals with MRKH syndrome have normal external genitalia, functioning ovaries, and a normal female karyotype (46, XX), they experience primary amenorrhea and are unable to carry a pregnancy. The existence of such severe developmental failure underscores the vital role the Müllerian ducts play in establishing the fundamental architecture of the female reproductive system.
Specific Müllerian Duct Anomalies
The detailed classification of Müllerian Duct Anomalies (MDA) provides a structured framework for understanding the precise embryological error involved. These defects are grouped according to the specific developmental stage that was compromised, allowing for targeted clinical assessment and treatment planning.
The ASRM classification system defines the major categories of Müllerian abnormalities:
- Class I: Failure of Formation (Agenesis or Hypoplasia): This includes MRKH syndrome, resulting in the complete absence or severe underdevelopment of the uterus and/or vagina.
- Class II: Failure of Fusion (Unicornuate Uterus): Involves the partial or complete failure of one Müllerian duct to develop, resulting in a uterus with only one functional horn and one fallopian tube. The absence of the contralateral duct is often associated with ipsilateral renal agenesis.
- Class III: Failure of Fusion (Uterus Didelphys): Characterized by the complete failure of the two ducts to fuse, resulting in two separate uteri, two cervices, and often a longitudinal vaginal septum.
- Class IV: Failure of Fusion (Bicornuate Uterus): Represents partial fusion failure, where the uterus is essentially heart-shaped, having two horns but a single cervix and lower uterine segment.
- Class V: Failure of Resorption (Septate Uterus): The most common type of MDA. Here, fusion occurred normally, but the central septum (the wall separating the two ducts prior to resorption) failed to dissolve completely, leaving a fibrous or muscular wall dividing the uterine cavity.
- Class VI: Failure of Resorption (Arcuate Uterus): Considered a minor anomaly, characterized by a slight indentation in the uterine fundus, representing a minimal failure of septal resorption, usually asymptomatic.
- Class VII: Combined Anomalies: Complex cases often involving exposure to teratogens, such as Diethylstilbestrol (DES), resulting in T-shaped uteri or other complex morphologies.
The clinical management of these anomalies varies dramatically. While septate uteri (Class V) can often be surgically corrected via hysteroscopic septoplasty to improve pregnancy outcomes, anomalies involving total agenesis (Class I) require alternative reproductive solutions, such as gestational surrogacy or uterine transplantation. The reproductive impact is highest in cases where the functional uterine cavity is compromised, such as the septate or unicornuate uterus.
Diagnostic Techniques and Management
Accurate diagnosis of Müllerian Duct Anomalies requires sophisticated imaging modalities, as the external genitalia are typically normal and the internal structures cannot be visualized through standard physical examination alone. The diagnostic journey often begins when an adolescent presents with primary amenorrhea or when an adult woman experiences recurrent miscarriage or infertility.
The primary diagnostic tools employed for comprehensive evaluation include:
- Pelvic Ultrasound: Provides an initial, non-invasive assessment of uterine size, shape, and the presence of two horns or fluid accumulation, though its resolution can be limited in complex cases.
- Hysterosalpingography (HSG): Historically used, this technique involves injecting contrast dye to outline the uterine cavity and fallopian tubes, primarily revealing septa or bicornuate configurations, though it is less accurate for determining the external contour of the uterus.
- Magnetic Resonance Imaging (MRI): Considered the gold standard, MRI provides superior soft-tissue contrast, allowing for precise visualization of the uterine external contour, the internal cavity, cervical structures, and the classification of the anomaly based on the musculature and fibrous tissue present.
- Laparoscopy and Hysteroscopy: These surgical procedures are used both for definitive diagnosis and correction. Laparoscopy confirms the external uterine contour, while hysteroscopy is essential for viewing and correcting intracavitary defects, such as the removal of a uterine septum (metroplasty).
Management strategies are highly individualized based on the specific anomaly and the patient’s symptoms and reproductive goals. For obstructive anomalies (e.g., transverse vaginal septum), surgical drainage is required to relieve pain and prevent endometriosis. For reproductive failures, surgical correction is aimed at creating a single, functional uterine cavity capable of sustaining a pregnancy. The goal of surgical intervention is always to normalize the anatomy as much as possible, thereby maximizing obstetric outcomes.
Genetic and Hormonal Regulation
The development, fusion, and ultimate fate of the Müllerian ducts are governed by a finely tuned network of genetic and hormonal signals, ensuring proper spatial and temporal control over differentiation. While the absence of AMH is the critical permissive factor for female development, the actual morphogenesis—the growth and shaping of the ducts—is regulated by various families of transcription factors.
Key genetic regulators include the Hox genes, specifically the HOXA group (e.g., HOXA10, HOXA11, HOXA13), which are vital master regulators that determine the regional identity along the reproductive tract axis. For instance, HOXA10 expression is crucial for uterine development, while HOXA13 is necessary for normal cervical and upper vaginal development. Mutations or misexpression of these genes are strongly linked to various types of Müllerian anomalies, demonstrating their foundational role in organogenesis.
Furthermore, the WNT signaling pathway plays a significant role in initiating and maintaining Müllerian duct development. WNT4, in particular, is essential for the initial establishment of the paramesonephric ducts and is implicated in suppressing the development of male characteristics in the female embryo. The combined interaction between genetic regulatory factors (HOX, WNT) and the endocrine environment (AMH, estrogens) dictates the final anatomical outcome, highlighting the intricate biological system necessary to produce a functional female reproductive tract from the initial bipotential embryonic structures.