Wildervanck Syndrome: Decoding a Rare Congenital Triad
- Introduction and Definition of Wildervanck’s Syndrome (Cervico-Oculo-Acoustic Syndrome)
- Etiology and Genetic Basis
- The Triad of Primary Clinical Manifestations
- Associated Skeletal and Craniofacial Abnormalities
- Neurological and Cognitive Features
- Differential Diagnosis and Correlation with Klippel-Feil Syndrome
- Epidemiology and Gender Predilection
- Management and Treatment Strategies
- Prognosis and Long-Term Outlook
Introduction and Definition of Wildervanck’s Syndrome (Cervico-Oculo-Acoustic Syndrome)
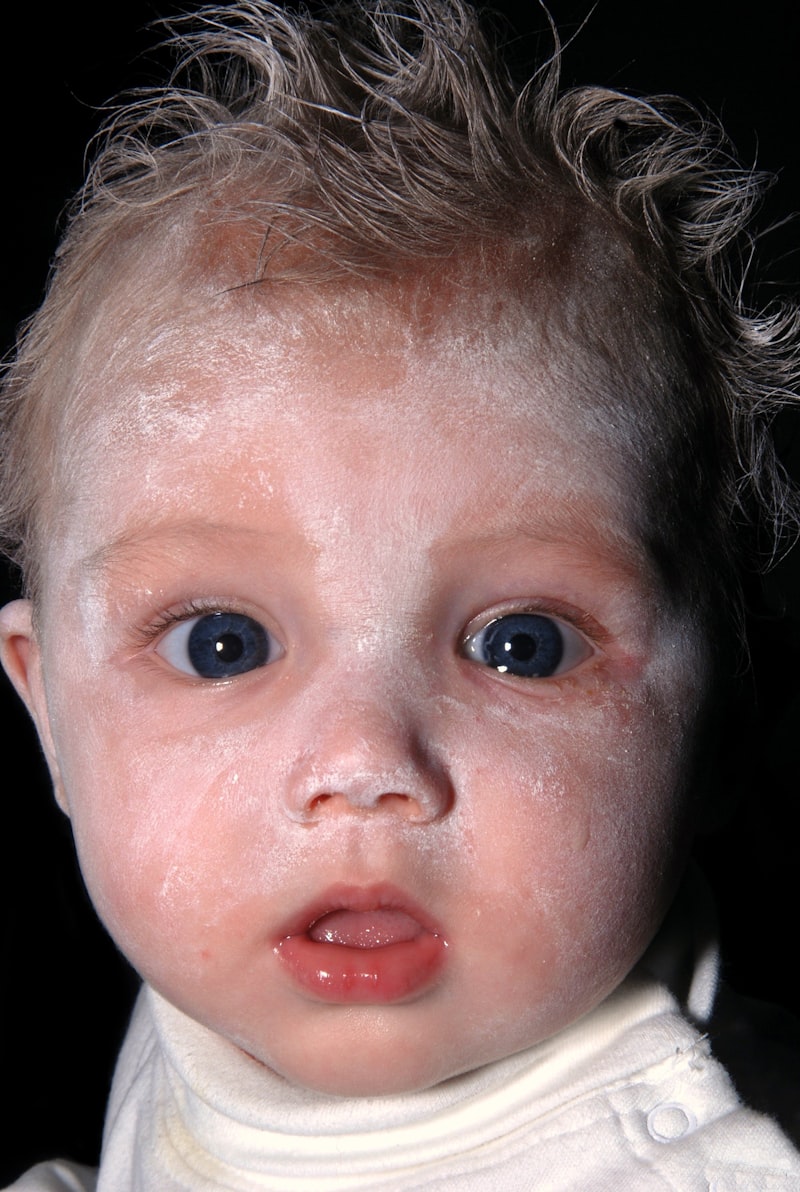
Wildervanck’s Syndrome, also formally recognized as Cervico-Oculo-Acoustic Syndrome, constitutes a complex and rare congenital disorder characterized by a distinct triad of symptoms involving the cervical spine, the eyes, and the auditory system. This condition is definitively classified as a genetic disorder, often presenting a significant overlap with the features of Klippel-Feil Syndrome (KFS), which primarily involves congenital fusion of two or more cervical vertebrae. The defining clinical presentation typically includes congenital deafness, ocular mobility impairment stemming from paralysis of the abducens nerve, and the characteristic skeletal anomalies associated with KFS. Recognition and early diagnosis are critical, as the constellation of symptoms can severely impact mobility, communication, and overall neurological development, necessitating a comprehensive, multidisciplinary approach to care throughout the patient’s lifespan. The syndrome was first thoroughly described by the Dutch ophthalmologist L.S. Wildervanck, highlighting the intricate interplay between disparate developmental failures originating during early embryonic stages, leading to these simultaneous malformations across multiple organ systems.
The description of Wildervanck’s Syndrome as a genetic disorder underscores its etiology rooted in developmental failures occurring before or during the first trimester of pregnancy. While the specific genetic mechanism remains elusive in many cases, its strong association with Klippel-Feil Syndrome suggests a shared or interrelated pathway concerning the proper segmentation and development of the somites, the structures that give rise to the vertebrae and associated musculature, and the neurodevelopmental structures of the inner ear and cranial nerves. The severity of the syndrome is highly variable among affected individuals, ranging from relatively mild manifestations where only one or two components of the triad are prominent, to severe, widespread involvement requiring extensive surgical and rehabilitative interventions. Therefore, understanding this syndrome requires appreciation for both the core diagnostic criteria and the broad spectrum of potential secondary complications, including those affecting cranial structure and cognitive function, which are not universally present but represent serious potential outcomes.
A key factor differentiating Wildervanck’s Syndrome from isolated congenital defects is the synchronous occurrence of specific cranial nerve dysfunction and vertebral anomalies. Specifically, the involvement of the abducens nerve (Cranial Nerve VI), which controls the lateral rectus muscle responsible for outward eye movement, resulting in abducens nerve paralysis (palsy), is a hallmark feature. This ocular impairment, combined with profound or severe hearing loss (deafness), sets it apart from simple Klippel-Feil presentations, establishing it as a distinct, yet overlapping, entity. Furthermore, the striking epidemiological pattern—where the condition is observed almost exclusively in the female population—provides essential clues regarding its underlying inheritance pattern, strongly suggesting an X-linked dominant trait that may be lethal in males, or another sex-linked mechanism influencing its expression.
Etiology and Genetic Basis
The precise etiology of Wildervanck’s Syndrome is categorized as a genetic disorder, though it typically follows a sporadic pattern or an uncertain mode of inheritance in individual families. Current research suggests that the condition likely arises from errors in early embryonic development, particularly during the critical period when the neural crest cells are migrating and differentiating, and when the segmentation of the paraxial mesoderm (leading to somites) is occurring. Given its strong correlation with Klippel-Feil Syndrome, which itself is heterogeneous but sometimes linked to genes such as GDF6 or GDF3 (Growth Differentiation Factor genes involved in skeletal development), it is hypothesized that Wildervanck’s Syndrome may represent a complex pleiotropic effect of mutations in genes governing multiple developmental pathways simultaneously, affecting both neurological structures (inner ear, abducens nucleus) and musculoskeletal elements (cervical vertebrae). The concurrent failure in the development of the cervical spine, the inner ear, and the abducens nucleus points toward a common, yet localized, developmental field defect.
Genetic studies have been complicated by the syndrome’s rarity and the significant gender bias. The observation that Wildervanck’s Syndrome occurs almost entirely in females suggests a compelling genetic hypothesis, most notably X-linked dominant inheritance. In this model, the defective gene residing on the X chromosome would be expressed in females (who possess two X chromosomes, allowing for survival), but could be lethal in utero for males (who possess only one X chromosome). This mechanism provides a robust explanation for the profound sex ratio distortion observed in clinical populations. However, the exact gene responsible for the full Wildervanck phenotype remains to be definitively identified, and researchers are continually exploring potential candidate genes involved in early neural and otic development, as well as those regulating the formation of the skull base and cranial nerves. The heterogeneity implies that while the clinical features converge into a recognizable syndrome, multiple distinct genetic mechanisms might ultimately lead to the same phenotypic presentation, complicating genetic counseling and specific diagnostic testing.
The developmental connection between the affected systems is crucial for understanding the disorder’s pathogenesis. The structures responsible for hearing (cochlea and semi-circular canals), the formation of the cervical spine, and the abducens nerve nucleus all develop in close spatial and temporal proximity during the fourth to eighth weeks of gestation. A localized insult, whether genetic or environmental, during this precise window could simultaneously disrupt all three processes. For instance, defective migration of the rhombomere-derived cells that form the abducens nucleus, coupled with abnormal formation of the otic capsule and segmentation failure in the cervical somites, provides a plausible embryological explanation for the characteristic triad. Future genetic sequencing efforts focusing on the X chromosome and genes critical for hindbrain patterning are essential to unlock the molecular basis of this challenging condition and provide targeted therapies or more accurate prognostic indicators based on genotype.
The Triad of Primary Clinical Manifestations
Wildervanck’s Syndrome is fundamentally defined by the presence of three major, interrelated clinical features, often referred to as a diagnostic triad. The first core element is congenital deafness, which is typically sensorineural in nature, although conductive or mixed hearing loss can also occur. The severity of the deafness is highly variable but frequently involves profound bilateral hearing loss, severely limiting speech and language development if not managed aggressively from infancy. This hearing impairment is often the result of malformations within the inner ear structures, specifically the cochlea and sometimes the acoustic nerve itself, reflecting the disruption of otic capsule development during embryogenesis. The presence of significant hearing loss necessitates early audiological evaluation and intervention, often involving hearing aids or cochlear implantation, to maximize auditory potential and communicative competence for the affected individual.
The second essential component of the triad is the specific ocular motility defect: paralysis of the abducens nerve (CN VI palsy). This palsy results in an inability to fully abduct (move outward) the affected eye, leading to esotropia (inward turning of the eye) and often causing diplopia (double vision) or compensatory head posturing to maintain binocular fusion. In some cases, the condition may manifest as Duane Retraction Syndrome, which is characterized by restricted abduction, retraction of the globe upon attempted adduction (inward movement), and narrowing of the palpebral fissure. The abducens nerve deficit is congenital and often attributed to hypoplasia or agenesis (underdevelopment or absence) of the abducens nucleus within the brainstem, or a malformation of the nerve itself as it attempts to innervate the lateral rectus muscle. Ophthalmologic intervention, including prisms or strabismus surgery, is frequently required to address the ocular misalignment and visual complications arising from this specific cranial nerve dysfunction.
The third critical manifestation is the presence of Klippel-Feil Syndrome (KFS), involving congenital fusion of at least two cervical vertebrae. This skeletal anomaly results in a short neck, a low posterior hairline, and a restricted range of motion in the neck. The severity of the KFS component varies dramatically; some patients exhibit only minor fusion, while others have extensive fusion of multiple segments, potentially leading to instability, chronic pain, and neurological compromise due to spinal cord compression. The presence of KFS requires careful orthopedic and neurosurgical monitoring, as the fused segments place undue mechanical stress on the unfused segments above and below, increasing the risk of premature degenerative changes and traumatic injury. The simultaneous presence of these three disparate, yet systematically linked, defects—auditory, ocular-neurological, and cervical-skeletal—solidifies the diagnosis of Wildervanck’s Syndrome and guides the extensive multidisciplinary management plan required for optimal patient care.
Associated Skeletal and Craniofacial Abnormalities
Beyond the mandatory presence of Klippel-Feil Syndrome (KFS) and its inherent cervical vertebral anomalies, patients with Wildervanck’s Syndrome frequently exhibit a wider range of associated skeletal and craniofacial abnormalities. These abnormalities often contribute significantly to the functional limitations and cosmetic concerns faced by affected individuals. The primary skeletal issues related to KFS include extensive fusion patterns in the cervical spine, but can also involve thoracic and lumbar segments. This fusion necessitates rigorous monitoring for secondary complications such as scoliosis or kyphosis, which develop as the spine attempts to compensate for the rigidity of the cervical region. The restricted neck mobility inherent to KFS can complicate daily activities and elevate the risk of injury during trauma, necessitating protective measures and regular orthopedic evaluations to maintain spinal stability and prevent progressive deformity.
A specific feature mentioned in the clinical description is the potential for cranial asymmetry. This asymmetry involves structural differences in the size and shape of the skull, often reflecting underlying developmental anomalies in the skull base and facial bones. Cranial asymmetry may be related to abnormal fetal positioning influenced by the stiff neck (torticollis secondary to KFS) or intrinsic defects in the ossification of the cranial vault. Furthermore, other craniofacial defects, such as cleft palate, facial nerve palsy (CN VII), or mandibular hypoplasia, have been reported in some cases, although they are not universally present. The presence of these structural anomalies underscores the widespread nature of the developmental disruption during embryogenesis, affecting tissues derived from multiple germ layers including the neuroectoderm and mesoderm.
Other less common, yet clinically significant, skeletal anomalies can include defects in the upper extremities, such as Sprengel deformity (congenital elevation of the scapula), and occasionally rib anomalies. These associated malformations further emphasize the systemic nature of the developmental defect in Wildervanck’s Syndrome, extending beyond the core triad. Management of these skeletal issues requires a collaborative approach involving pediatric orthopedics and physical therapy to maximize function, minimize pain, and address potential long-term complications stemming from structural imbalances. Early identification and proactive intervention regarding these widespread bony malformations are crucial for improving the patient’s long-term physical capacity and quality of life, particularly regarding complex activities involving fine and gross motor skills.
Neurological and Cognitive Features
The neurological component of Wildervanck’s Syndrome is prominently defined by the congenital abducens nerve paralysis, which is a specific form of cranial neuropathy. This involvement of Cranial Nerve VI highlights a fundamental disruption in the central nervous system development, specifically concerning the brainstem nuclei responsible for ocular motor control. The neurological impact extends beyond the eye movement disorder, however, as the profound sensorineural deafness suggests damage or maldevelopment of the auditory pathway, either at the level of the inner ear receptors or the auditory nerve itself. The interdependence of these neurological and sensory deficits means that affected individuals face significant challenges in spatial orientation, balance (due to vestibular involvement often associated with inner ear defects), and primary communication acquisition.
An additional, though variable, neurological feature documented in the syndrome is the potential for cognitive retardation. While not a universal feature, the potential for intellectual disability requires thorough developmental screening and assessment in all diagnosed cases. When cognitive deficits are present, they can range from mild learning difficulties requiring specialized educational support to more profound intellectual impairments. The mechanism underlying cognitive involvement is not fully understood but may relate to generalized cerebral malformations, hydrocephalus (excess fluid in the brain), or secondary effects stemming from chronic sensory deprivation (hearing loss) and communication deficits. The variability in cognitive outcomes suggests that secondary factors or co-existing, yet unidentified, genetic modifiers might influence the degree of intellectual function achieved.
Furthermore, a subset of patients may exhibit other central nervous system anomalies, including syringomyelia (cysts within the spinal cord), tethered cord syndrome, or structural abnormalities of the cerebellum or corpus callosum. These secondary neurological findings necessitate advanced imaging studies, such as Magnetic Resonance Imaging (MRI), to fully delineate the extent of CNS involvement. The management of these neurological features is highly individualized, focusing on early intervention programs, specialized education tailored to the child’s specific cognitive and sensory profile, and, when necessary, neurosurgical procedures to address conditions like hydrocephalus or spinal cord compression. Addressing both the sensory deficits and potential cognitive impairments simultaneously is paramount for fostering optimal development and maximizing adaptive functioning throughout life.
Differential Diagnosis and Correlation with Klippel-Feil Syndrome
The diagnosis of Wildervanck’s Syndrome is established clinically through the recognition of its characteristic triad, but it necessitates careful differentiation from other conditions that share some of its features. The most crucial differential diagnosis involves distinguishing it from isolated Klippel-Feil Syndrome (KFS) or isolated congenital deafness syndromes. While Wildervanck’s Syndrome is inherently correlated with KFS—meaning KFS is a mandatory diagnostic component—the addition of the specific ocular (abducens nerve palsy) and auditory (deafness) deficits transforms the diagnosis. Simple KFS, while sharing the cervical spinal fusion, typically lacks the consistent, profound involvement of the cranial nerves and inner ear observed in Wildervanck’s Syndrome. Therefore, the presence of combined cervico-ocular-acoustic anomalies is the distinguishing criterion that moves the diagnosis from KFS alone to the more complex Wildervanck’s classification.
Other conditions that must be excluded include various forms of congenital cranial dysinnervation disorders (CCDDs), such as Möbius Syndrome, which involves paralysis of multiple cranial nerves, often CN VI and CN VII (facial nerve). Although Möbius Syndrome can also present with abducens palsy and sometimes other midline defects, it usually lacks the defining Klippel-Feil spinal anomalies and the characteristic severe inner ear deafness seen in Wildervanck’s Syndrome. Furthermore, congenital hearing loss syndromes, such as Waardenburg syndrome or Pendred syndrome, must be considered, but these conditions do not typically feature the combination of vertebral fusion and specific CN VI palsy. The comprehensive clinical assessment, including audiometry, detailed ophthalmological examination, and high-resolution imaging (X-rays and MRI) of the cervical spine and brainstem, is essential to correctly classify the patient’s condition and rule out overlapping yet distinct congenital disorders.
The strong correlation between Wildervanck’s Syndrome and Klippel-Feil Syndrome suggests a shared genetic susceptibility or a common pathway of developmental disruption affecting the segmentation of the somites. Many researchers consider Wildervanck’s Syndrome to be a severe, specialized subtype or spectrum disorder within the broader category of Klippel-Feil related conditions, where the genetic mutation has a wider pleiotropic effect encompassing structures derived from the adjacent neural plate and otic placode. This inherent connection dictates that all individuals diagnosed with Wildervanck’s Syndrome must undergo rigorous orthopedic screening for the full spectrum of KFS-related complications, including occult spinal instability, kidney anomalies (which commonly co-occur with KFS), and potential neurological deterioration stemming from cervical cord compression, ensuring that the management plan addresses both the cranial and spinal elements of the disease.
Epidemiology and Gender Predilection
A defining and highly significant epidemiological feature of Wildervanck’s Syndrome is its pronounced gender predilection. The condition occurs almost entirely in females, making it one of the most striking examples of sex-biased congenital syndromes. While extremely rare case reports involving males exist, they are often considered exceptions or potentially atypical variants, reinforcing the overwhelming clinical observation that the vast majority of diagnosed individuals are female. This extreme skew in the sex ratio provides critical insight into the probable genetic mechanisms underlying the disorder, strongly pointing toward an X-linked dominant mode of inheritance. In such a scenario, a mutation on the X chromosome would be dominant in females (who have two X chromosomes) but would likely be lethal to male fetuses (who have only one X chromosome, known as hemizygosity), resulting in early pregnancy loss or stillbirths, thus explaining the virtual absence of affected males in the live-born population.
The true prevalence of Wildervanck’s Syndrome is difficult to ascertain precisely due to its rarity and the potential for underdiagnosis, particularly in cases where the KFS component is subtle or the deafness is mild. However, based on reported clinical series, it is considered one of the rarest congenital disorders, often diagnosed only by highly specialized medical centers focusing on complex craniofacial and neuro-otological anomalies. The rarity emphasizes the highly specific and likely severe nature of the developmental insult required to simultaneously disrupt the formation of the cervical spine, the inner ear, and the abducens nucleus, all while adhering to a strict sex-linked pattern of survival. Genetic counseling for families affected by Wildervanck’s Syndrome must carefully consider this X-linked dominant hypothesis, particularly when assessing recurrence risk for future pregnancies, focusing on the significantly higher risk for female offspring and the likely lethality for male offspring.
Understanding the female predominance is not merely an academic exercise; it impacts clinical management and genetic research strategies. The disproportionate representation of females necessitates a focus on gender-specific health concerns that may arise in this population, though the primary clinical challenges remain tied to the core triad of symptoms regardless of sex. Ongoing genetic research often focuses on identifying mutations on the X chromosome that could account for this pattern, potentially leading to the discovery of a master regulatory gene involved in the complex patterning of the hindbrain and mesoderm during the crucial weeks of embryonic development. The highly specific sex ratio remains the most compelling clue guiding the search for the underlying pathogenic locus of this rare and complicated syndrome.
Management and Treatment Strategies
The management of Wildervanck’s Syndrome demands a highly coordinated, multidisciplinary treatment plan due to the complexity and diversity of the affected systems. Treatment is primarily symptomatic and supportive, aiming to mitigate the functional deficits arising from the congenital anomalies. A core component of the management team includes specialists from audiology, ophthalmology, orthopedics, neurosurgery, and developmental pediatrics. Early intervention is critical, particularly for addressing congenital deafness, which requires prompt audiological assessment and the provision of appropriate amplification devices, such as hearing aids, or surgical options like cochlear implants, to facilitate optimal language acquisition and development. Educational specialists must be involved early to implement strategies for communication, whether through spoken language supported by amplification or alternative methods like sign language.
The ocular manifestations, particularly the abducens nerve paralysis, require specialized ophthalmological care. Treatment often involves corrective lenses, prisms to manage minor degrees of strabismus, or surgical correction of the extraocular muscles (strabismus surgery) to improve alignment, reduce diplopia, and address compensatory head postures. Regular monitoring is essential to prevent the development of amblyopia (lazy eye) secondary to the visual misalignment. Orthopedic management focuses on the Klippel-Feil component, requiring periodic radiographic assessments of the cervical spine to monitor for instability, progressive fusion, or the development of secondary scoliosis or kyphosis. Physical therapy is often employed to maintain the maximum possible range of motion and prevent muscle contractures, while patients must be educated on the inherent risks associated with neck trauma due to the underlying vertebral fragility and fusion.
Furthermore, any associated neurological symptoms, such as the potential for cognitive retardation or structural CNS anomalies (e.g., syringomyelia), require specialized care. Developmental pediatricians and neurologists oversee cognitive assessments and coordinate access to special education services, physical therapy, and occupational therapy to address developmental delays and maximize adaptive skills. Given the potential for renal or cardiac anomalies often associated with KFS, a comprehensive systemic workup, including renal ultrasound and echocardiogram, is typically recommended upon diagnosis. The overall goal of the comprehensive management plan is to proactively address the wide spectrum of potential complications, stabilize existing functional deficits, and empower the individual to achieve the highest possible level of independence and quality of life through continuous, coordinated care tailored to the specific needs dictated by the severity of the congenital defects.
Prognosis and Long-Term Outlook
The prognosis for individuals diagnosed with Wildervanck’s Syndrome is highly variable and depends intrinsically on the severity of the core triad of symptoms and the presence of associated anomalies, particularly the degree of cognitive involvement and spinal instability. In cases where the deafness is manageable with cochlear implants or high-power hearing aids, the abducens palsy is surgically corrected, and the Klippel-Feil fusion is stable without significant neurological compromise, individuals can lead relatively independent and fulfilling lives, often achieving normal educational and professional milestones. Early diagnosis and the aggressive initiation of multidisciplinary intervention, especially concerning auditory and visual rehabilitation, are paramount determinants of favorable long-term outcomes.
Conversely, the outlook is more guarded for individuals presenting with profound, uncorrectable sensorineural deafness, severe, intractable ocular motility deficits, or extensive cervical fusion leading to chronic spinal instability or spinal cord compression. The presence of significant cognitive retardation is the factor that most substantially limits long-term independent functioning, requiring lifelong supportive care and specialized living arrangements. Patients must also be monitored throughout adulthood for progressive spinal deterioration, including accelerated disc degeneration above and below the fused segments, which may necessitate future surgical interventions to manage pain or prevent neurological injury. Therefore, continuous orthopedic and neurological surveillance is a necessity, extending well beyond childhood.
Despite the challenges inherent in managing a complex, congenital syndrome, advances in surgical techniques (both orthopedic and ophthalmological) and prosthetic devices (cochlear implants) have significantly improved the functional capacity and quality of life for many affected individuals. Psychological and social support are also crucial components of the long-term outlook, helping patients and families cope with the chronic nature of the condition, managing communication barriers, and fostering self-advocacy. While Wildervanck’s Syndrome presents a lifetime of management complexities, a proactive, patient-centered, and interdisciplinary approach offers the best pathway toward maximizing development, minimizing long-term disability, and ensuring integration into the community.