Aniridia: Seeing Beyond the Missing Iris
- Definition and Scope of Aniridia
- Etiology and Genetic Basis
- Ocular Manifestations and Primary Symptoms
- Visual Impairment and Prognosis
- Secondary Ocular Complications: Glaucoma and Cataracts
- Systemic Associations: WAGR Syndrome and Ataxia
- Diagnosis and Management Strategies
- Psychological and Developmental Impact
Definition and Scope of Aniridia
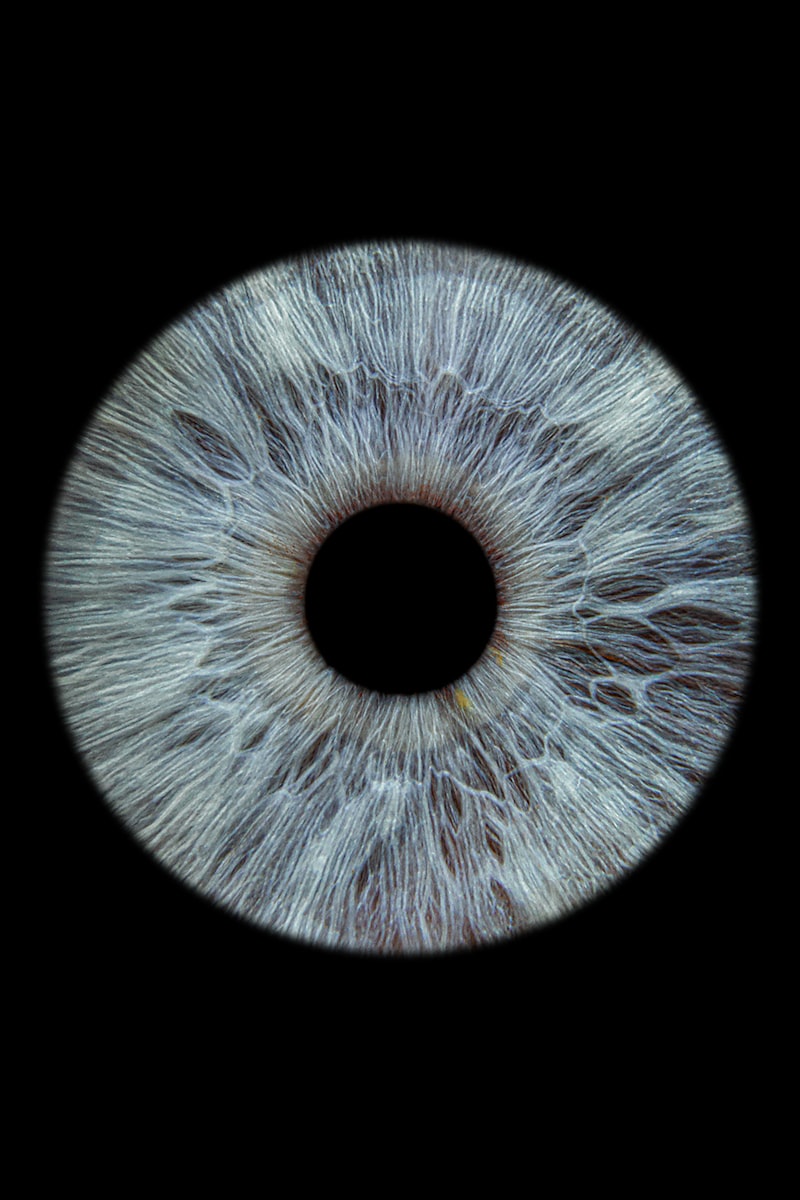
Aniridia, derived from the Greek meaning “without iris,” is an extremely rare panocular congenital disorder characterized primarily by the complete or partial absence of the iris, the structure responsible for regulating the amount of light entering the eye. This condition is not merely a cosmetic defect; it represents a complex developmental failure affecting multiple ocular tissues, leading to significant visual impairment and a high probability of severe secondary complications. While the absence of the iris is the most recognizable feature, the pathology of aniridia extends far deeper, often involving concurrent hypoplasia or malformation of other critical structures necessary for clear sight, including the macula lutea and the optic nerves. This broad spectrum of underlying structural deficiencies necessitates a comprehensive understanding of the disorder beyond its superficial appearance, placing it within the category of severe congenital ocular diseases with profound neurodevelopmental implications.
The prevalence of aniridia is often cited as approximately 1 in 50,000 to 100,000 live births globally, underscoring its classification as an ultra-rare disease. Although some cases may present sporadically, the condition is most frequently inherited through an autosomal dominant pattern, emphasizing the critical role of genetic mutation in its etiology. Phenotypically, aniridia can range from a near-complete absence of the iris tissue, resulting in a large, dark pupil-like aperture, to a subtle hypoplasia where only vestigial remnants of the iris are present. This variability in presentation dictates the immediate visual consequences and influences the trajectory of secondary conditions that almost inevitably arise. The foundational problem, however, remains the inability to properly control light exposure, leading to severe photophobia and degraded image quality on the retina, even if the lenses and corneas initially appear structurally sound.
Etiology and Genetic Basis
The primary genetic driver of aniridia is a mutation or deletion in the PAX6 gene, located on chromosome 11p13. This gene is a crucial transcription factor, often referred to as a master control gene, essential for the development of the eye, central nervous system, and pancreas. Mutations in PAX6 disrupt the complex cascade of events required for the proper formation of the anterior segment of the eye during embryogenesis, specifically affecting the development of the iris stroma, retina, lens, and cornea. Typically, aniridia follows an autosomal dominant inheritance pattern, meaning a child only needs to inherit one copy of the mutated gene from either parent to develop the condition. Approximately two-thirds of cases are familial, while the remaining one-third arise from sporadic mutations, often linked to larger chromosomal deletions that may encompass neighboring genes, leading to more complex systemic syndromes.
Understanding the molecular mechanism is paramount to appreciating the full scope of the disorder. The PAX6 protein is vital for regulating the expression of numerous downstream genes that orchestrate the differentiation and migration of cells forming the iris and other anterior segment structures. When this regulatory function is impaired, the resulting tissue hypoplasia is irreversible. Furthermore, the severity of the mutation—whether it is a nonsense mutation leading to a truncated, non-functional protein or a missense mutation altering function—can correlate with the overall clinical presentation. Genetic testing is therefore a critical component of diagnosis, not only confirming the presence of the PAX6 mutation but also helping to distinguish isolated aniridia from syndrome-associated forms, particularly those involving large deletions that extend into the neighboring WT1 gene, a scenario characteristic of WAGR syndrome.
Ocular Manifestations and Primary Symptoms
Beyond the conspicuous absence of the iris, aniridia involves a constellation of related ocular anomalies that collectively contribute to profound visual deficit. The iris tissue that is present is often fibrovascular remnants or thin, underdeveloped stroma, unable to function effectively as a diaphragm to control light entry. This leads to debilitating photophobia, or extreme light sensitivity, as the retina is constantly exposed to excessive and uncontrolled illumination. Another almost universal finding is nystagmus, an involuntary, rhythmic movement of the eyes. Nystagmus arises due to the underlying foveal and macular hypoplasia, where the fovea, the central point of sharpest vision, fails to develop properly, preventing the establishment of stable fixation reflexes necessary for precise visual tracking.
Crucially, the poor visual prognosis is intrinsically linked to the hypoplasia of the macula lutea and the optic nerves. The macula and fovea are underdeveloped, meaning the specialized cones required for high-resolution central vision are absent or poorly organized. Similarly, the optic nerve, which transmits visual information from the retina to the brain, may exhibit hypoplasia, being smaller and having fewer functional axons than normal. These structural defects in the posterior pole of the eye are independent of the iris defect but are genetically related via the PAX6 expression pattern. It is these posterior segment anomalies, rather than the iris defect alone, that are the primary determinants of the patient’s final visual acuity, which is typically severely limited, often stabilizing around 20/200 or worse.
Visual Impairment and Prognosis
Patients diagnosed with aniridia consistently face severe, lifelong visual impairment. The expected level of sight is generally quantified around 20/200, placing individuals within the legal definition of blindness in many jurisdictions. This level of acuity means that what a person with normal vision can see clearly at 200 feet, the person with aniridia must be 20 feet away to perceive with the same clarity. However, the functional impact extends beyond simple acuity measures. The constant presence of photophobia, coupled with nystagmus, makes daily activities requiring fine motor skills, tracking moving objects, or navigating brightly lit environments exceptionally challenging. The absence of a functional iris severely reduces contrast sensitivity and depth perception, further complicating visual tasks.
The long-term prognosis for vision is often complicated by the progressive nature of secondary complications. While the initial structural abnormalities set the baseline for poor sight, the subsequent development of glaucoma and cataracts acts as a secondary layer of visual degradation. Therefore, the management strategy must focus not only on optimizing the use of the remaining sight through low-vision aids and environmental modifications but also on aggressively monitoring and treating these progressive secondary diseases, which pose the greatest threat to the preservation of any residual vision. Early intervention and consistent ophthalmological surveillance are absolutely critical to preventing further irreversible sight loss.
Secondary Ocular Complications: Glaucoma and Cataracts
Two of the most serious and common secondary complications arising in aniridia patients are cataracts and glaucoma, developing in nearly all affected individuals over time. Cataracts, the clouding of the lens, typically manifest early in life, often requiring surgical intervention. The mechanism is believed to be related to the underlying PAX6 dysfunction affecting lens development and metabolism. While cataract surgery can improve clarity, the outcomes are often guarded due to the pre-existing poor macular and foveal function, meaning that the removal of the cataract does not restore vision to normal levels but merely clears the central visual axis.
Glaucoma, characterized by damage to the optic nerve usually due to elevated intraocular pressure (IOP), represents perhaps the most threatening complication to residual vision. The prevalence of glaucoma in aniridia patients approaches 50% to 75%, and it often presents resistant to typical medical management. The cause is structural: the abnormal development of the anterior segment angle, the drainage pathway for aqueous humor, results in a congenital obstruction or malformation known as aniridic glaucoma. This blockage leads to a chronic increase in IOP, which progressively damages the already compromised optic nerve. Early detection and aggressive management—often requiring filtering surgery or the placement of drainage devices—are essential to delay or prevent total blindness caused by irreversible glaucomatous optic atrophy.
Systemic Associations: WAGR Syndrome and Ataxia
While isolated aniridia is primarily an ocular disorder, approximately one-third of sporadic cases are linked to larger chromosomal deletions on 11p13, resulting in systemic conditions. The most well-known of these is WAGR syndrome, an acronym for its defining features: Wilms’ tumor (a kidney cancer), Aniridia, Genitourinary abnormalities, and intellectual Retardation (developmental delay). WAGR syndrome occurs when the deletion encompasses not only the PAX6 gene but also the adjacent WT1 gene, which is a tumor suppressor. The presence of WAGR syndrome drastically alters the patient’s prognosis, necessitating multidisciplinary care focusing on oncology and developmental pediatrics in addition to ophthalmology.
Furthermore, the clinical description often links aniridia to a specific neurological presentation sometimes termed Aniridia-Ataxia Syndrome, which aligns with the systemic involvement noted in the defining text. This latter syndrome is characterized by significant neurological deficits, including mental retardation (developmental delay or intellectual disability), severe ataxia (lack of muscle control and coordination), and difficulties with speech articulation (dysarthria) and forming words. These neurological manifestations highlight the wide-ranging developmental role of the PAX6 gene and its associated pathways in central nervous system development. The co-occurrence of severe ocular pathology and profound neurological impairment mandates specialized educational and therapeutic interventions focused on motor skill development, speech therapy, and managing intellectual challenges.
Diagnosis and Management Strategies
Diagnosis of aniridia is typically made shortly after birth based on the clear physical appearance of the absent or rudimentary iris. Confirmation involves a detailed ophthalmological examination using a slit lamp to assess the extent of the iris hypoplasia, foveal hypoplasia, and potential involvement of the optic nerve head. Crucially, genetic testing for PAX6 mutations is indispensable, particularly to differentiate between isolated familial cases and sporadic cases that may indicate the presence of WAGR syndrome. If WAGR syndrome is suspected, screening for Wilms’ tumor through regular abdominal ultrasound monitoring is immediately initiated, often continuing through the first eight years of life, underscoring the necessity of integrated care upon diagnosis.
Management of aniridia is complex, multi-faceted, and focused primarily on minimizing secondary damage and maximizing the use of residual vision. Because the condition is genetic and irreversible, treatment is palliative rather than curative. Key management strategies include the use of tinted contact lenses or prosthetic iris implants to reduce photophobia and improve cosmesis. However, these optical interventions must be balanced against the increased risk of corneal complications, a common issue in aniridia known as keratopathy. Pharmacological management often involves aggressive control of intraocular pressure to prevent glaucoma progression, although surgical intervention, such as trabeculectomy or tube shunt placement, is frequently required due to the resistance of aniridic glaucoma to medical therapies. Additionally, low-vision specialists are essential to provide aids, magnifiers, and environmental adjustments tailored to the patient’s specific level of acuity and functional challenges.
Psychological and Developmental Impact
The profound visual impairment, coupled with the potential for systemic neurological involvement, places significant psychological and developmental burdens on individuals with aniridia and their families. Early onset of severe visual loss interferes directly with crucial developmental milestones, particularly those related to motor skills, spatial awareness, and non-verbal communication, often requiring specialized early intervention programs. The need for frequent medical appointments, surgical procedures, and lifelong monitoring introduces chronic stress and anxiety. From a psychological perspective, children and adolescents must navigate social environments, including school, while managing visible differences (the appearance of the eye), photophobia, and the functional restrictions imposed by legal blindness, necessitating robust emotional and social support systems.
For those individuals associated with systemic syndromes like WAGR or Aniridia-Ataxia, the challenges are further compounded by intellectual and physical disabilities. The requirement for specialized education, physical therapy to address ataxia and poor muscle control, and speech therapy for difficulties forming words places immense demands on family resources and support structures. Psychosocial interventions must address issues of self-esteem, coping mechanisms related to chronic illness, and the development of independent living skills adapted for severe visual and physical limitations. Successful long-term outcomes depend heavily on the seamless integration of ophthalmological care with developmental psychology, educational resources, and genetic counseling.