Cranial Bifida: Understanding Rare Developmental Traits
Introduction and Defining Characteristics
Cranial Bifida, a rare and highly specific developmental disorder, is fundamentally defined by its primary clinical manifestation: a distinct, symmetrical impression in the center of the forehead. This indentation is unique in its morphology, consistently presenting in a characteristic horseshoe shape, often described in clinical texts as an inverted arch or a U-shaped depression situated along the midline of the frontal bone. Unlike many congenital skull irregularities that result from trauma or generalized syndromes, Cranial Bifida is classified primarily as a hereditary disorder, suggesting a specific genetic predisposition influencing the ossification and fusion processes of the cranial structures during early fetal development. The disorder is typically identified immediately following parturition, as the structural anomaly is visually and palpably evident, confirming its status as a congenital condition. The severity of the impression can vary along a spectrum, ranging from a slight, superficial groove requiring careful tactile examination, to a profound, deep depression that significantly alters the contour of the forehead, thereby forming the basis for clinical grading and prognosis determination.
The core interest in Cranial Bifida stems from its singular presentation, which differentiates it from broader conditions affecting craniofacial development, such as craniosynostosis or various forms of meningocele, where structural abnormalities are often accompanied by severe neurological deficits or widespread systemic involvement. In contrast, Cranial Bifida frequently presents as an isolated structural anomaly, though its presence necessitates careful screening for potential underlying soft tissue or dural involvement, given the proximity of the impression to the inner table of the frontal skull. The clinical literature emphasizes that while the physical manifestation is striking, the associated functional impairment, if any, must be thoroughly assessed, placing a heavy burden on initial diagnostic protocols to rule out secondary complications related to cerebral protection or underlying vascular anomalies that might co-exist with the bony defect. The enduring nature of the horseshoe impression means that affected individuals carry this marker throughout their lives, making understanding the long-term psychosocial integration of this permanent physical trait a crucial aspect of longitudinal care.
Furthermore, the term bifida implies a division or splitting, which, in this context, refers not to a complete failure of midline closure as seen in spina bifida, but rather to a localized failure of the complete fusion or robust development of the frontal midline suture, resulting in the characteristic depressed arch. This failure is genetically programmed, manifesting reliably across generations within affected families, which has allowed researchers to trace complex inheritance patterns, though the precise locus of the responsible gene or gene cluster remains a subject of intensive investigation. The clinical observation that the anomaly is evident from the moment of birth underscores the importance of prenatal screening and genetic counseling for families with a known history of the condition. Understanding the precise anatomical layers affected—skin, subcutaneous tissue, periosteum, and the underlying frontal bone—is essential for both accurate diagnosis and the planning of any potential corrective or reconstructive surgical interventions that might be deemed necessary based on the depth of the impression or the patient’s cosmetic concerns.
Historical Context and Nomenclature
The recognition of Cranial Bifida as a distinct clinical entity evolved slowly through medical history, often confused with traumatic injuries or localized dermatological atrophy due to its unique indentational morphology. Early descriptions, dating back to 19th-century European anatomical texts, referred vaguely to “frontal depressions” or “coronal clefts,” but lacked the specificity regarding the horseshoe shape that now defines the condition. It was not until the early 20th century, with the advent of standardized radiographic techniques, that the bony origin of the impression could be definitively confirmed, differentiating it from purely superficial soft tissue anomalies. The formal nomenclature, Cranial Bifida, was adopted to highlight the hypothesized embryological root—a localized defect in the midline integration of the frontal bone—while emphasizing its location within the cranium. This naming convention helped to move the condition out of the realm of general craniofacial malformations and into a specialized category focusing on hereditary bony defects.
The primary challenge in establishing a clear historical record lies in the fact that, frequently, individuals presenting with Cranial Bifida exhibited no associated neurological or cognitive deficits, meaning the condition was often cataloged as a purely cosmetic variation rather than a medical pathology requiring aggressive intervention. This minimized its visibility in mortality and morbidity statistics, delaying focused research efforts into its etiology. However, detailed genealogical studies conducted in specific isolated communities, where the hereditary pattern was particularly pronounced, proved instrumental in demonstrating the condition’s Mendelian inheritance pattern, solidifying its classification as a hereditary disorder. These studies meticulously tracked the appearance of the characteristic impression in the center of the forehead through multiple generations, providing the strongest evidence that the condition was not sporadic but rather genetically transmitted.
The historical evolution of treatment methodologies reflects the changing perceptions of the disorder. Initially, interventions were non-existent or focused solely on camouflaging the defect with hairstyles or cosmetic fillers. As surgical techniques advanced, particularly in the mid-to-late 20th century, attempts were made to reconstruct the frontal bone using grafts or synthetic materials, though such procedures were reserved for the most severe cases where the depression was significantly deep or caused secondary issues related to sinus drainage or increased vulnerability to minor head trauma. The contemporary approach, informed by decades of longitudinal follow-up, tends toward minimal intervention unless functional issues are present, recognizing the benign nature of the anomaly in the majority of cases. The consistent documentation that the symptom is evident from the moment of birth has allowed for retrospective analysis of neonatal records, providing a wealth of standardized data that supports current diagnostic protocols, ensuring immediate identification of the condition in newborns.
Etiology and Genetic Basis
As a confirmed hereditary disorder, the etiology of Cranial Bifida is rooted in a highly specific genetic mechanism that disrupts the intricate developmental timeline of the frontal skeletal structure. Current hypothesis centers around a mutation affecting genes responsible for the signaling pathways governing intramembranous ossification of the frontal bone plates. The frontal bone originates from two separate halves which normally fuse at the metopic suture, a process typically completed entirely within the first two years of life, though the initial stages of approximation occur prenatally. In Cranial Bifida, the genetic defect appears to cause a localized hypoplasia or insufficient bone deposition along the superior aspect of this developmental midline, resulting in the characteristic symmetrical depression. This failure is not a complete absence of bone, but rather a localized thinning and backward bowing, which manifests externally as the horseshoe shape impression.
Genetic studies have utilized linkage analysis in large affected families to narrow down potential chromosomal regions. While no single causative gene has been universally identified across all pedigrees, research strongly suggests an autosomal dominant inheritance pattern with variable penetrance. The concept of variable penetrance is essential for understanding why the structural anomaly, though always present, can vary significantly in depth and prominence—from a faint cosmetic line to a deeply recessed groove. Furthermore, some affected individuals may carry the responsible allele but display such a minimal external sign that they are historically classified as unaffected, complicating accurate genetic counseling and epidemiological tracking. The consistency of the presentation—always the impression in the center of the forehead—suggests that the mutation targets a key regulatory element active only during the specific window of frontal bone morphogenesis.
The embryological timing of the defect is critical: because the anomaly is evident from the moment of birth, the developmental error must occur relatively early in the second trimester, when the foundation for the frontal bone is being rapidly laid down. A related theory proposes that the defect might not solely be one of bone formation, but potentially involves the dura mater—the tough protective membrane surrounding the brain. An abnormal adherence or tethering of the dura to the inner aspect of the developing frontal plates could exert a localized tension, mechanically drawing the bone inward and contributing to the arching, depressed morphology. This hypothesis accounts for the unique symmetry and depth of the impression, and also necessitates high-resolution imaging studies (such as MRI) in diagnostics to evaluate the relationship between the bony defect and the underlying neurovascular structures, although serious neurological involvement is rare.
Clinical Presentation and Diagnostics
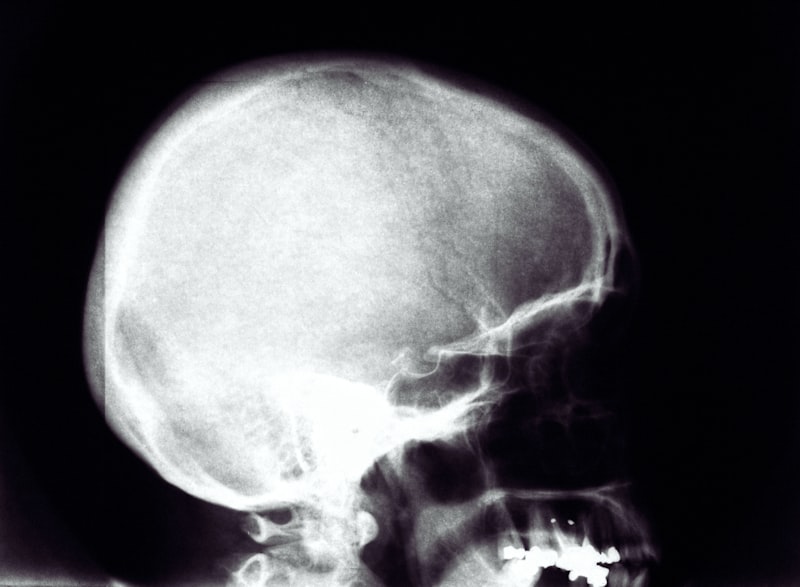
The clinical presentation of Cranial Bifida is dominated by the palpable and visible impression in the center of the forehead. This depression is typically situated just superior to the nasion and extends upward towards the hairline, forming a distinct, inverted U or horseshoe shape. The size and depth are the most variable clinical parameters. In mild cases, the impression may be subtle, only visible when the patient raises their eyebrows or when examined under tangential lighting, and may feel like a slight groove upon palpation. In severe presentations, the depression can be several millimeters deep, creating a prominent indent that casts a visible shadow and significantly alters the profile of the patient’s brow ridge. Crucially, the skin overlying the depression is typically normal in appearance, texture, and coloration, distinguishing it from syndromic conditions associated with overlying skin atrophy or vascular malformations.
The diagnostic process begins with a meticulous physical examination immediately following birth, since the condition is evident from the moment of birth. If the horseshoe impression is identified, the next critical step involves radiological confirmation of the underlying bony defect. Plain film radiography may show a localized area of thinning or bowing of the frontal bone, but modern diagnostics strongly favor advanced cross-sectional imaging, primarily Computed Tomography (CT) scans, which provide superior visualization of the osseous structure. High-resolution CT allows clinicians to precisely measure the depth of the depression, assess the thickness of the remaining bone, and crucially, determine the proximity of the defect to the frontal sinuses and the anterior cranial fossa. This detailed anatomical mapping is vital for risk assessment and surgical planning, should intervention be considered.
Further diagnostic investigation often includes Magnetic Resonance Imaging (MRI), particularly when there is a suspicion of underlying soft tissue involvement, or if the impression is exceptionally deep. MRI is indispensable for evaluating the integrity of the dura mater, assessing for any associated dural sinuses anomalies, or ruling out an occult meningocele—a rare complication where cranial membranes protrude through a bony defect. While Cranial Bifida is generally an isolated bony defect, thorough diagnostic screening is necessary to ensure the absence of associated congenital anomalies, given its classification as a hereditary disorder. Clinicians must also gather a detailed family history, constructing a pedigree chart to confirm the hereditary pattern and inform genetic counseling, noting the presence of the horseshoe shape anomaly in first- and second-degree relatives.
Differential Diagnosis
Distinguishing Cranial Bifida from other craniofacial anomalies is paramount, given its specific genetic etiology and generally benign prognosis compared to more complex disorders. The primary diagnostic distinction lies in the unique morphology—the horseshoe shape localized strictly to the frontal midline—and its lack of systemic involvement. Conditions that must be ruled out include minor forms of Metopic Craniosynostosis, where premature fusion of the metopic suture leads to a pointed, triangular forehead (trigonocephaly). While both involve the metopic suture area, Craniosynostosis involves premature *fusion* leading to ridging, whereas Cranial Bifida involves localized *hypoplasia* resulting in a depressed impression. Furthermore, Craniosynostosis can restrict brain growth, a complication rarely associated with Cranial Bifida.
Another key differential is the presence of a congenital dermoid cyst or a localized lipoma in the frontal area. These soft tissue masses can sometimes cause pressure remodeling of the underlying bone, mimicking a depression. However, these masses typically present as swelling or a palpable subcutaneous nodule, whereas the impression associated with Cranial Bifida is solely structural—a defect of the bony contour itself, with normal overlying soft tissue. Imaging (ultrasound, CT, or MRI) is decisive in separating these entities, confirming whether the depression is caused by an intrinsic bony anomaly or extrinsic soft tissue pressure. Moreover, the strong pattern of familial transmission in this hereditary disorder provides a crucial historical distinction that is usually absent in sporadic dermoid cyst presentations.
Finally, traumatic deformities, particularly those sustained during birth or infancy, must be excluded, though the symmetrical, arching nature of the Cranial Bifida impression is highly characteristic and usually inconsistent with localized blunt force trauma. The defining characteristic that helps eliminate trauma as a cause is the fact that the condition is evident from the moment of birth, documented immediately in neonatal records. Other rare considerations include localized forms of osteomalacia or nutritional rickets, though these are typically generalized conditions affecting the entire skeleton and are easily ruled out through blood work and generalized skeletal surveys. The specificity of the location and the horseshoe shape remain the key identifiers that lock in the diagnosis of Cranial Bifida.
Psychosocial Impact and Management
While Cranial Bifida is typically medically benign, the permanent visibility of the impression in the center of the forehead necessitates careful consideration of its significant psychosocial impact. For many affected individuals, particularly adolescents and young adults, the unique frontal contour can lead to self-consciousness, body image issues, and social anxiety. The forehead is a highly exposed and central feature of the face, and having a consistently visible structural anomaly, even if purely cosmetic, can affect peer relationships and overall self-esteem. Management, therefore, extends beyond the medical and surgical realms, requiring robust psychological support and counseling, starting early in childhood to foster healthy coping mechanisms and self-acceptance regarding the hereditary disorder.
Management strategies are generally individualized based on the depth of the defect and the patient’s level of distress. For mild cases, non-invasive approaches are prioritized. These include education for the patient and family, emphasizing the lack of associated brain pathology, and psychological intervention aimed at normalization and resilience building. For patients experiencing severe distress or those with very deep indentations, reconstructive surgery may be an option. Surgical intervention typically involves cranioplasty techniques, utilizing bone cement (methyl methacrylate) or customized PEEK (polyether ether ketone) implants shaped specifically to fill the horseshoe shape defect and restore the smooth contour of the frontal bone. These procedures are complex and carry inherent risks, and are generally delayed until facial growth is complete to minimize the need for revision.
A crucial component of early management involves detailed genetic counseling for the parents and, eventually, the affected individual, particularly since the condition is strongly classified as a hereditary disorder. Counseling must clearly explain the mode of inheritance, the variable penetrance, and the probability of transmission to future offspring. Furthermore, given that the symptom is evident from the moment of birth, immediate parental support is vital. Parents must be educated to understand that the structural difference does not imply cognitive delay or severe functional impairment, helping them to frame the diagnosis positively and advocate for their child’s psychological well-being throughout their development. Long-term follow-up studies confirm that while the visible difference is permanent, most individuals with Cranial Bifida achieve normal psychosocial integration with appropriate support.
Prognosis and Longitudinal Studies
The long-term prognosis for individuals diagnosed with Cranial Bifida is overwhelmingly positive, especially concerning neurological function and overall quality of life. The condition is generally characterized as a structural anomaly with minimal or no functional impairment, distinguishing it favorably from other congenital craniofacial defects. Longitudinal studies, some spanning several decades, have tracked cohorts of individuals presenting with the characteristic impression in the center of the forehead and confirmed that the bony defect is typically static, meaning it does not progress or worsen over time once the initial growth period is complete. Furthermore, the risk of secondary medical complications related to the indentation, such as increased susceptibility to infection or trauma, remains exceptionally low.
These extensive longitudinal investigations have also provided critical data regarding the stability of the hereditary pattern. By tracking multiple generations, researchers have confirmed that the underlying genetic mechanism remains stable, consistently producing the defining horseshoe shape impression, though the variable penetrance continues to account for the spectrum of severity observed. Crucially, these studies have been essential in definitively separating Cranial Bifida from conditions that might share a superficial resemblance but carry a risk of late-onset complications, thereby reinforcing the current clinical consensus that aggressive neurosurgical monitoring is typically unwarranted unless specific indications arise from initial advanced imaging. The primary concern tracked in long-term follow-up remains the psychological adjustment of the patient to the permanent visible marker of the hereditary disorder.
Regarding surgical outcomes, studies assessing the efficacy of reconstructive cranioplasty procedures indicate high patient satisfaction rates, provided the patient’s primary motivation is cosmetic improvement and they have realistic expectations regarding the outcome. While surgery can successfully restore a smooth frontal contour, it is important to note that the underlying genetic predisposition remains unchanged, and careful monitoring for any potential implant-related issues is necessary throughout the patient’s life. Overall, the consistent finding that the condition is evident from the moment of birth, coupled with decades of benign follow-up data, allows clinicians to reassure families that Cranial Bifida, while unique in presentation, does not typically compromise health or lifespan.
Modern Research and Future Directions
Contemporary research into Cranial Bifida is focused intensely on molecular genetics, aiming to isolate the specific gene or genes responsible for the hereditary disorder. Advances in high-throughput sequencing technologies, such as whole-exome sequencing, are currently being applied to large, multi-generational pedigrees where the condition is prevalent. The goal is to identify a reliable genetic marker that can provide precise prenatal diagnosis and deepen the understanding of the developmental pathways that lead to the localized hypoplasia of the frontal bone. Identifying the causative mutation would allow for highly accurate risk assessment in genetic counseling, moving beyond empirical recurrence risks based on population data.
Another key area of investigation involves advanced biomechanical modeling of the frontal bone development. Researchers are using computational fluid dynamics and finite element analysis to simulate the stresses and growth patterns within the developing fetal cranium. The aim is to understand exactly how the hypothesized genetic defect translates into the physical manifestation of the horseshoe shape depression. This modeling seeks to determine if the impression is purely a result of insufficient bone deposition, or if subtle, abnormal forces exerted by the dura or underlying brain tissue contribute to the characteristic arching shape of the defect. Such insights could potentially lead to highly targeted, non-surgical interventions, perhaps involving early fetal therapy or specialized cranial molding techniques, although these remain highly theoretical at present.
Future directions also include the development of standardized, objective metrics for classifying the severity of the impression in the center of the forehead. While current classification relies heavily on subjective visual assessment and basic CT measurements, the integration of 3D photogrammetry and quantitative surface analysis tools will allow clinicians to precisely grade the depth, volume, and symmetry of the defect. This standardized approach will facilitate more rigorous clinical trials regarding reconstructive techniques and allow for better correlation between the physical severity of the anomaly and the patient’s reported psychosocial burden. As Cranial Bifida is a condition evident from the moment of birth, continued research into early detection and personalized intervention strategies remains a priority within specialized craniofacial centers globally.