Craniosynostosis: Understanding Early Skull Development
- Introduction: Defining Craniostenosis and Its Implications
- Anatomical Basis and Normal Cranial Development
- Classification and Types of Craniostenosis
- Etiology and Underlying Risk Factors
- Clinical Manifestations and Diagnostic Procedures
- Impact on Neurocognitive Development
- Treatment Modalities and Surgical Intervention
- Long-Term Prognosis and Management
Introduction: Defining Craniostenosis and Its Implications
Craniostenosis, often referred to as craniosynostosis, is a critical pediatric condition characterized by the premature fusion of one or more cranial sutures. These sutures are fibrous joints that connect the bony plates of the skull, which are typically designed to remain open and flexible during infancy and early childhood. This flexibility is essential for accommodating the rapid growth of the underlying brain structures. The fundamental issue in craniostenosis is that this early fusion restricts the skull’s ability to expand perpendicularly to the fused suture, leading to a distinctive and often severe malformation of the skull shape, along with potentially devastating neurodevelopmental consequences.
The impact of this premature closure extends far beyond cosmetic concerns regarding head shape. Since the human brain triples its size during the first two years of life, any constraint upon this expansion places undue pressure on the delicate neural tissues. This mechanical restriction limits the average growth of the brain’s constructs, preventing the brain from achieving its full volumetric potential. The resulting constraint can elevate intracranial pressure (ICP), which, if sustained and untreated, can compromise neuronal function and myelination. Therefore, craniostenosis is not merely a skeletal defect but a significant neurological risk factor that necessitates prompt intervention to mitigate long-term functional impairment.
While the original content noted that craniostenosis “occurs post-birth when pieces of a child’s skull begin to fuse together,” it is more accurate to state that the pathological process begins either prenatally or perinatally, becoming clinically apparent at or shortly after birth. The consequences of this restricted growth are severe, frequently manifesting as cognitive retardation and various levels of developmental delay. Understanding the intricate relationship between cranial volume restriction and compromised cognitive development forms the core of its study within neurodevelopmental psychology. The severity of the cognitive outcome is often correlated with the number of sutures involved and the timeliness of surgical correction, emphasizing the need for early diagnosis and specialized multidisciplinary care.
Anatomical Basis and Normal Cranial Development
To fully grasp the pathology of craniostenosis, it is crucial to first understand the normal anatomy and developmental timeline of the infant skull. The neurocranium is composed of several distinct bony plates—frontal, parietal, temporal, and occipital—which are separated by the aforementioned sutures. Key sutures include the sagittal suture (running front-to-back along the top of the head), the coronal sutures (separating the frontal bone from the parietal bones), the lambdoid sutures (separating the parietal bones from the occipital bone), and the metopic suture (running down the center of the forehead). These sutures are functionally designed as growth centers, allowing the skull vault to expand in response to the growing brain.
In a typical development pathway, these sutures remain patent (open) throughout the first several years of life, providing crucial plasticity. The brain undergoes its most explosive period of growth between birth and 24 months, increasing dramatically in size and complexity. The open sutures facilitate this expansion, and their premature closure—the definition of craniostenosis—immediately dictates the direction of restricted growth. If a suture fuses, growth is halted perpendicular to that suture line, forcing compensatory growth parallel to the fused suture. This leads to the characteristic, often bizarre, shapes associated with different types of the condition.
Furthermore, the anatomy includes the fontanelles, commonly known as soft spots, which are areas where multiple sutures intersect (e.g., the anterior and posterior fontanelles). While the fontanelles provide important clinical markers, it is the activity and patency of the linear sutures that primarily govern skull shape and volume expansion. The timing of normal closure is highly specific; for instance, the metopic suture typically closes relatively early (around 3 to 9 months), while the sagittal and coronal sutures remain open much longer. Craniostenosis occurs when this physiological timeline is dramatically accelerated, often resulting in complete bony bridging years before it should naturally occur, severely compromising the potential for maximum brain development.
Classification and Types of Craniostenosis
Craniostenosis is generally classified based on the specific suture or combination of sutures that prematurely fuse. These classifications are critical because the fused suture determines the resulting head shape, the potential for elevated intracranial pressure, and the specific surgical approach required for correction. The most common form is Scaphocephaly, resulting from the early fusion of the sagittal suture. This restriction inhibits lateral expansion, forcing the skull to grow long and narrow (boat-shaped). Scaphocephaly accounts for approximately 40–60% of all non-syndromic cases, and while it often has a less severe impact on ICP compared to multi-sutural fusions, it still requires corrective action.
Another significant type is Coronal Craniostenosis, which involves the premature closure of one or both coronal sutures. Unilateral coronal fusion leads to Anterior Plagiocephaly, causing a flattening of the forehead on the affected side and compensatory bulging on the opposite side. Bilateral coronal fusion results in Brachycephaly, where the skull is abnormally short and wide due to restricted growth from front to back. Brachycephaly is often associated with a higher risk of increased intracranial pressure because it restricts growth across a major axis, directly impacting the volume available for the frontal and parietal lobes.
Less common but equally important forms include Trigonocephaly, caused by the fusion of the metopic suture, which results in a triangular shape to the forehead with a ridge running down the midline. Lambdoid Craniostenosis, involving the fusion of one or both lambdoid sutures, leads to posterior flattening (Posterior Plagiocephaly). Furthermore, complex or multi-suture craniostenosis, where three or more sutures fuse simultaneously, presents the most challenging clinical picture. These cases are highly correlated with syndromic forms (such as Apert, Crouzon, or Pfeiffer syndromes) and carry the highest risk for severe neurocognitive deficits and persistent, dangerously elevated intracranial pressure, demanding comprehensive and often multi-stage surgical intervention.
Etiology and Underlying Risk Factors
The causes of craniostenosis are broadly categorized into two main groups: primary (non-syndromic) and secondary (syndromic). Primary craniostenosis, which accounts for the vast majority of cases (approximately 80–90%), is generally considered idiopathic, meaning the specific cause is unknown. However, research suggests a multifactorial etiology involving both genetic predisposition and localized biomechanical factors. For instance, single-suture synostoses like Scaphocephaly are often sporadic, though slight familial tendencies have been observed, suggesting subtle genetic influences that have not been fully elucidated.
Secondary, or syndromic, craniostenosis is linked to identifiable genetic mutations and structural syndromes. These conditions often involve multiple suture fusions and frequently present with associated skeletal, facial, and limb abnormalities. Key syndromes include Apert syndrome, characterized by severe synostosis and complex syndactyly (fused fingers or toes); Crouzon syndrome, which involves midface hypoplasia and shallow orbits leading to bulging eyes; and Pfeiffer syndrome, which includes broad thumbs and toes. These syndromic forms are often caused by mutations in specific genes, particularly the Fibroblast Growth Factor Receptor (FGFR) genes (e.g., FGFR1, FGFR2, and FGFR3). These genetic aberrations disrupt the signaling pathways responsible for regulating bone growth and differentiation, leading directly to the premature ossification of the cranial sutures.
Beyond intrinsic genetic factors, several extrinsic or environmental risk factors have been implicated, although their definitive causal role remains debated. These include maternal exposures during pregnancy, such as certain anticonvulsant medications (e.g., valproate), maternal hyperthyroidism, and mechanical forces exerted on the fetal head during gestation, particularly in cases of uterine constraint or oligohydramnios. While these factors may not directly cause the condition, they might predispose genetically susceptible infants to early suture closure. Ultimately, a combination of heritable genetic mutations and subtle biomechanical or environmental triggers likely contributes to the heterogeneous presentation of this cranial malformation, complicating preventative strategies.
Clinical Manifestations and Diagnostic Procedures
The most obvious clinical manifestation of craniostenosis is the severely abnormal head shape, which is usually noticeable at birth or shortly thereafter and progresses rapidly as the brain attempts to grow. The specific morphology—long and narrow (scaphocephaly), wide and short (brachycephaly), or triangular (trigonocephaly)—provides the initial clue to the involved sutures. Physical examination often reveals a palpable, bony ridge running along the line of the fused suture, which is firm and non-mobile, contrasting sharply with the soft, pliable nature of open sutures.
Beyond the morphological changes, the most critical clinical signs relate to elevated intracranial pressure (ICP), which occurs when the restricted skull volume can no longer accommodate the growing brain and normal cerebrospinal fluid dynamics. While ICP is less common in isolated, single-suture synostosis (except perhaps bilateral coronal), it is a major concern in multi-sutural and syndromic cases. Symptoms of increased ICP in infants can be subtle and non-specific, including irritability, persistent vomiting, lethargy, bulging fontanelle (if still open), and dilated scalp veins. In older children, classic symptoms such as intractable headaches, visual disturbances (due to papilledema), and developmental regression become more pronounced.
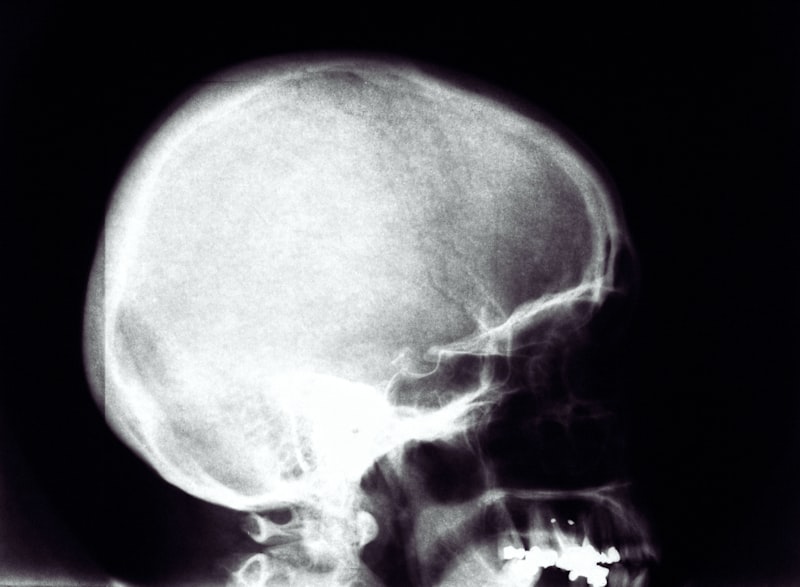
Diagnosis relies heavily on advanced imaging techniques. Initial screening may involve skull X-rays, which can clearly reveal the absence of a suture line and the presence of bony bridging. However, the gold standard for definitive diagnosis, surgical planning, and assessment of brain structures is a High-Resolution Computed Tomography (CT) scan with 3D reconstruction. The CT scan accurately maps the extent of the suture fusion, identifies potential secondary signs of increased ICP (such as ‘beaten copper’ appearance on the inner table of the skull), and provides surgeons with the detailed spatial information required for planning complex cranial vault reconstruction. Early and accurate diagnosis is essential, as the window for optimal surgical intervention is narrow, typically occurring before the child reaches one year of age.
Impact on Neurocognitive Development
The most profound and concerning aspect of craniostenosis, particularly relevant to psychology and neurodevelopment, is its potential to cause long-term neurocognitive impairment. The mechanical restriction of brain growth, coupled with chronic or intermittent elevation of intracranial pressure, directly interferes with the normal processes of neuronal migration, synaptogenesis, and cerebral blood flow. When the brain is physically constrained, specific cortical regions may not develop their full functional capacity. Studies consistently show that children with untreated or complex craniostenosis have a significantly higher incidence of developmental delays compared to the general population.
These deficits are often observed across multiple domains. Cognitive function, including Intelligence Quotient (IQ) scores, frequently falls below average, particularly in cases involving multiple fused sutures or syndromic presentations. Beyond generalized cognitive delays, specific learning disabilities are common, often involving executive function deficits, attention problems (ADHD), and difficulties in spatial reasoning and mathematical skills. Furthermore, the psychosocial aspect cannot be ignored; the visible disfigurement and the need for multiple complex surgeries can lead to significant psychological stress, impacting self-esteem and social integration, which further complicates overall developmental trajectories.
The correlation between specific suture involvement and cognitive outcomes is complex but informative. While isolated sagittal synostosis (scaphocephaly) generally carries a lower risk for severe intellectual disability compared to conditions like bilateral coronal synostosis (brachycephaly), all forms require careful developmental monitoring. Early diagnosis and timely surgical decompression are the primary protective factors against severe neurocognitive sequelae. Surgical intervention aims not only to correct the skull shape but, more importantly, to normalize intracranial pressure and provide the necessary volume for the brain to resume unrestricted growth, thus maximizing the potential for typical cognitive development. Lifelong specialized educational and psychological support is often necessary, even after successful surgical correction.
Treatment Modalities and Surgical Intervention
The primary treatment for nearly all forms of symptomatic craniostenosis is surgical correction, which aims to release the fused sutures, reshape the cranial vault, and, critically, increase the intracranial volume to prevent or resolve elevated pressure. The timing of surgery is paramount, as procedures performed during the peak period of brain growth (ideally between 3 and 12 months of age) tend to yield the best long-term outcomes, particularly concerning neurodevelopmental prognosis. Surgical approaches vary significantly based on the child’s age, the number of sutures involved, and the presence of syndromic features.
For infants under six months with single-suture synostosis, minimally invasive techniques are often preferred. These include endoscopic strip craniectomy, where a narrow strip of the fused bone is removed endoscopically through small incisions. This technique is less invasive and requires shorter hospital stays but mandates the use of postoperative helmet therapy (cranial orthosis) for several months to guide and maintain the corrected skull shape. For older infants or those with complex, multi-sutural synostosis, more extensive open procedures, known as Cranial Vault Remodeling (CVR), are necessary. CVR involves large incisions, the removal and reshaping of large sections of the skull bone, and their re-plating using resorbable fixation materials to create a normalized cranial shape and adequate volume.
In severe syndromic cases, such as those involving significant midface retraction (e.g., Crouzon syndrome), complex procedures utilizing advanced techniques like Distraction Osteogenesis (DO) may be employed. DO involves surgically cutting the bone and applying specialized devices to gradually separate the bone fragments over several weeks or months. This slow, controlled expansion allows new bone to fill the gap, providing maximum volume gain. Regardless of the technique chosen, surgical correction is a high-stakes procedure requiring specialized neurosurgical and craniofacial teams. The ultimate goal remains the relief of restricted growth and intracranial pressure to ensure the best possible functional and cosmetic outcomes, minimizing the risk of permanent brain damage and cognitive impairment.
Long-Term Prognosis and Management
The long-term prognosis for children with craniostenosis is highly variable and depends predominantly on the initial severity, the presence of associated syndromes, and the timing and effectiveness of surgical intervention. For children with isolated, single-suture synostosis who undergo successful early surgery, the prognosis is generally excellent, with most achieving normal cognitive development and requiring minimal subsequent intervention beyond routine follow-up. However, these children still require careful monitoring for specific learning difficulties or psychosocial adjustments, as the condition represents a significant early life trauma.
Conversely, children diagnosed with syndromic craniostenosis or those with multi-sutural involvement face a more guarded prognosis. These individuals often require multiple revision surgeries throughout childhood and adolescence to manage recurrent or persistent suture fusion, address midface hypoplasia, and correct orbital issues. Critically, the management of these complex cases must extend beyond the surgical realm to encompass comprehensive developmental pediatric, neurological, and psychological support. Persistent elevated intracranial pressure remains a life-long risk in some syndromic conditions, requiring vigilant ophthalmological and neurological assessments.
A key component of long-term management involves addressing the identified cognitive and developmental delays. Formal neuropsychological testing is essential to identify specific areas of weakness, such as issues with spatial awareness, fine motor skills, or executive function. Based on these findings, individualized educational programs and targeted therapies (physical therapy, occupational therapy, speech therapy) are implemented to maximize functional abilities. Furthermore, psychological support is vital for both the child and the family to navigate the challenges associated with complex medical journeys, visible differences, and the intensive demands of chronic medical management, ensuring the child can achieve the highest possible quality of life despite their underlying condition.